Primers for a Simple Identification
of Genetically Modified Soya (GTS) in Food by PCR
Lain Mencia Martinez, Christian Lozanoski,
Dietmar Scherr, Max-Beckmann-Schule, Oberstufengymnasium
in Frankfurt am Main, Sophienstr. 70, D-60487 Frankfurt
Abstract
Three PCR primer pairs for the specific, sensitive and rapid detection
of genetically modified Soya (Glycine max) are described here. Soya
containing the EPSPS gene (5-enolpyruvyl-3-phosphoshikimate synthase, EC
2.5.1.19 ) of bacterial origin /1/, /2/ (Agrobacterium
sp. strain designated
CP4 ), generating a glyphosate tolerance
of soya plants, can be detected by three pairs of primers resulting in
a 388 bp (bp = base pair), or alternatively a 267 bp or a 227 bp amplicon.
Key words Genetically modified soya, PCR
Primers
Introduction
The detection of genetically modified Soya in food will be of great
interest for all who are involved in surveying soya containing products /3/,
/4/.Primers are
described here that work very well under the simple conditions of a school
laboratory, using a very slow thermocycler (Pharmacia LKB Gene ATAG Controller).
The extraction of the genomic DNA of the samples which is essential for
a qualified detection was performed by standard procedures (Promega). The
primers were developed for a nested PCR but which is not necessary (Fig.
2). The primers yielding a 227 bp amplicon could be useful when DNA in
food is very degradated.
Materials and methods
To avoid contamination the extractions and PCR were performed in different
parts of the laboratory. Extractions, as well as the tubes and instruments
for mincing were handeled in containments of plastic.
Soya seed was soaked at 65 °C for 1 h before extraction.
As an example for food containing genetically modified soya (soya protein)
we describe here experiments with Nestle Salvimulsin MCT "Sondennahrung".
The suspension was directly extracted.
Extraction
200 mg of the minced soya seed sample or 300 µl of the suspension
(Nestle Sondennahrung) was mixed with 500 µl of extraction buffer
(10 mM Tris-HCl, 150 mM NaCl, 2mM EDTA, w = 1% SDS), 50 µl 5 M Guanidin-
HCl solution (Serva), 20 µl of Proteinase K (Boehringer Mannheim)
(20mg/ml). The tube was inverted and incubated for at least 1 h at 57 °C,
followed by a centrifugation for 10 min at 10000 x g at room temperature.
The solution was added to 1 ml of Wizard DNA purification resin (Promega,
no. A 767C) and the Promega clean-up procedure was performed. The DNA was
eluted with 80 °C heated TE buffer (Tris-HCl, pH 7.5, 1 mM EDTA)
Preparation of standards
DNA extracts of genetically modified seed was diluted 1 : 10, 1 : 100,
1 : 1000, 1 : 3000 and 1 : 10000 with extracts of unmodified seed. We estimated
the concentration of DNA in those extracts by gel electrophoresis to yield
about 1 µg/µl. 1 µl of the diluted sample 1 : 1000 contains
1µg unmodified DNA and 1 ng modified DNA.
Primers
The primers were constructed for positions on the 3' end of the CP4
EPSPS gene (Sense) and on the 5' end of the NOS gene (antisense).
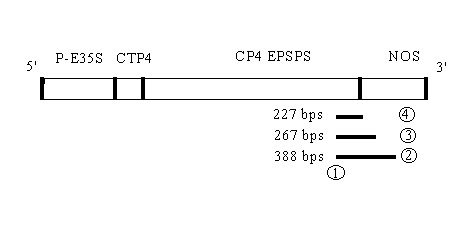
Figure 1: Integrated fragment: P-E35S Promoter, CTP4: N-terminal chloroplast
transit peptid sequence from Petunia Hybrida EPSPS gene, CP4
EPSPS: bacterial EPSPS gene, NOS: 3' nontranslated region of the nopaline
synthase gene (terminator)
The position of the primers: sense 1 and different antisense primers
2, 3, 4 and the length of the amplicons (bp):
Sense primer
1 CP4 EPSPS
5' CATCTCGATCACCGCATCGCCATG 3'
Antisense primers
2 NOS 3'
5' CGTATTAAATGTATAATTGCGG 3'
3 NOS 3'
5` CGCAAGACCGGCAACAGGATTC 3'
4 NOS 3'
5' TGCCAAATGTTTGAACGATC 3'
PCR and DNA analysis
PCR was carried out in a total volume of 50 µl containing 5 µl
10 X buffer I (15 mM Mg2+) (Perkin Elmer), 1 µl BSA (Serva) (2mg/ml),
4 µl dNTP (5mM), 5 µl primer sense (1 µM), 5 µl
primer antisense, 24.5 µl A.dem., 5 µl template, 0.5 µl
Taq Polymerase (Perkin Elmer). Two drops of mineral oil was added.
First denaturation (hotstart) at 94 °C for 5 min, then Taq polymerase
was added. 40 cycles were performed at 95 °C 30 s, 53 °C 30 s and
72 °C 60 s, final extension 72 °C 5 min.
DNA analysis was performed with AluI yielding a visible restriction
product of 204 bp. The 388 bp fragment was sequenced confirming the results
obtained by restriction analysis.
Discussion
The performance of PCR utilizing the above listed
primers and the Promega extraction procedure is qualified for identification
of GTS in food, as we have shown for Nestle "Sondennahrung" (Fig. 2, Fig.
4). The sensitivity is enough to identify in a sample 300 pg template DNA
beside 1 µg unmodified Soya DNA. (Fig. 3)

Figure 2: Agarose gel electrophoresis of a PCR of genetically modified
soya (GTS ) lane 1: 100 base pair ladder (Gibco); lane 2: negative control (without
template); lane 3: soya seed DNA extract (primer 1 and 2); lane 4: soya
seed DNA extract (primer 1 and 3); lane 5: GTS extract (primer 1 and 2);
lane 6: GTS extract (primer 1 and 3); lane 7: nested PCR; lane 8: GTS extract
(primer 1 and 4)lanes 9-13 PCR, applying different conditions
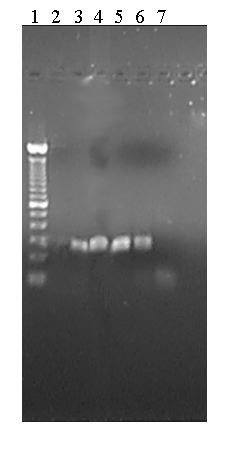
Figure 3: Sensitivity test, lane 1: 100 base pair ladder (Gibco), lane
2: negative control, without template, lane 3: GTS 1: 10 diluted with soya
DNA extract, lane 4: 1:100 ; lane 5: 1:1000 ; lane 6: 1:3000 ; lane 7:
1 : 10000
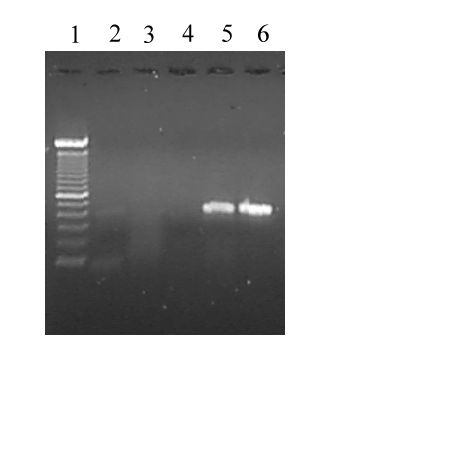
Figure 4: 100 bp ladder (Gibco), lane 2: Negative control, without
template, lane 3: nonmodified soya seed DNA extract, lane 4: soya flour
DNA extract ; lane 5: GTS DNA extract; lane 6: Nestle Sondennahrung DNA
extract
Literature:
/1/ United States Patent, G.F. Barry et al.,
Monsanto Company, St Louis, Mo., Patent Number 5,633,435 Date of Patent
May 27, 1997
/2/S.R. Padgette
et al., Crop Sci. 35:1451-1461 (1995)
/3/E. Köppel
et. al., Mitt. Gebiete Lebnsm Hyg. 88, 164-175 (1997)
/4/J.A. Straub et
al., Z Lebensm Unters Forsch A (1999) 208: 77-82